What is MEGA?
Molecular Evolutionary Genetics Analysis or MEGA is a sophisticated and user-friendly software suite for analyzing DNA and protein sequence data. Here I will show how it can be used for sequence alignments and inferring phylogenies.
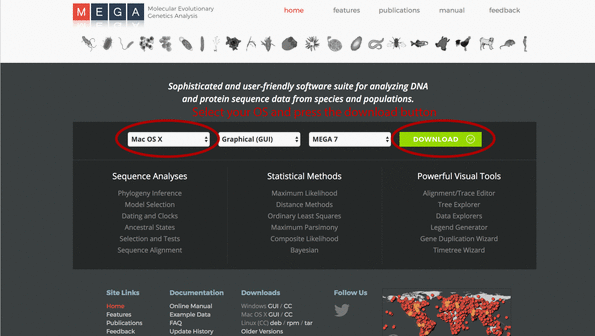
1. Go to the MEGA homepage and select your operating system, press the download button, and install the program to your computer.
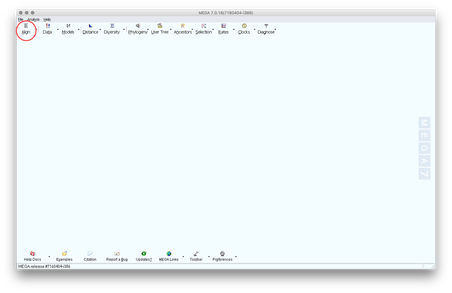
2. Lauch MEGA.
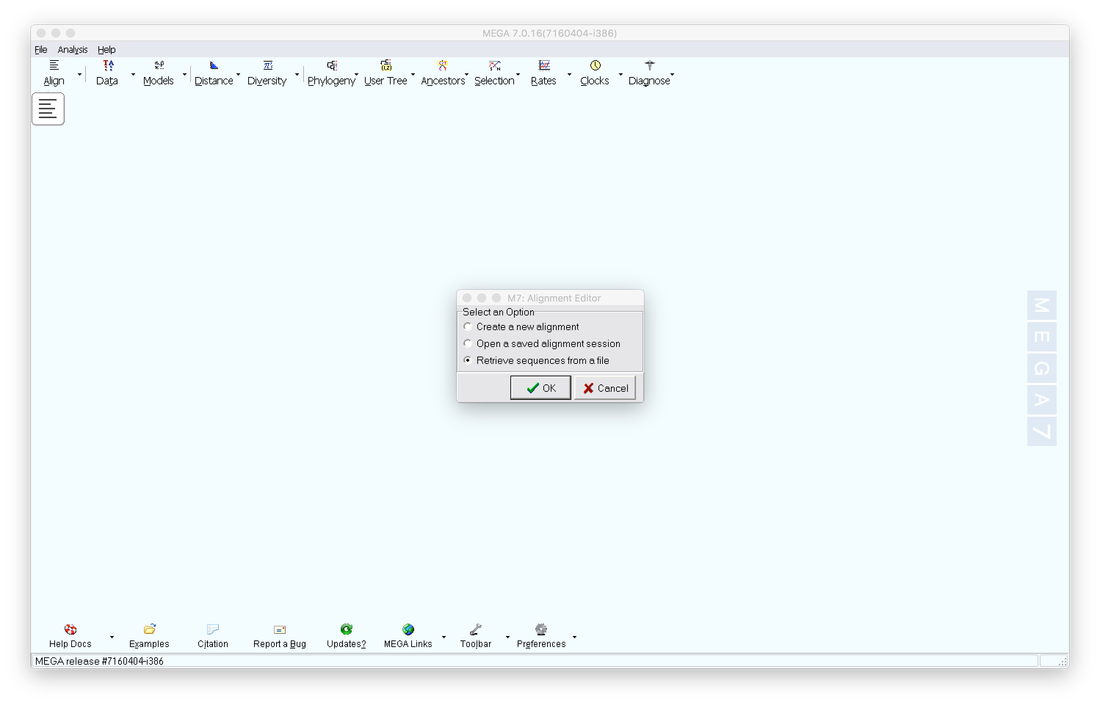
3. Open your FASTA file in MEGA. Click on the Align tab, then select Edit/Build Alignment, lastly select Retrieve sequences from a file.
|
|
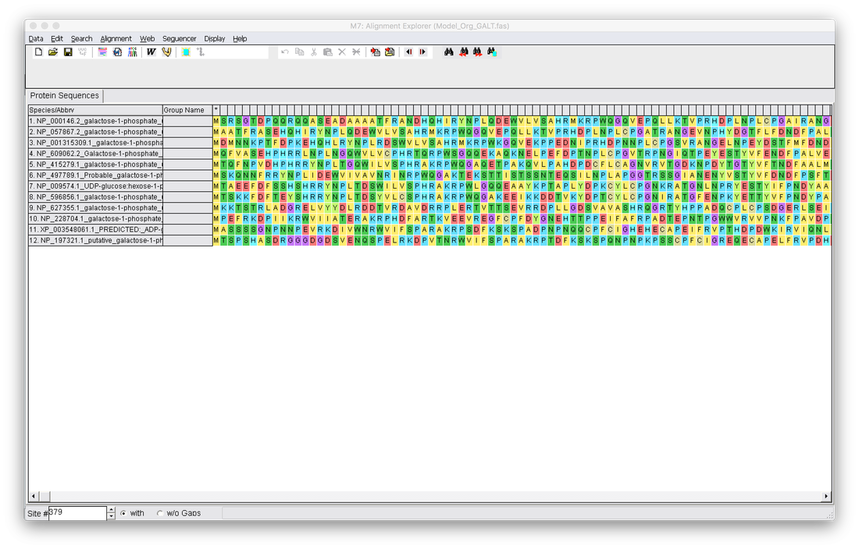
4. Once you have loaded your FASTA file into mega you will see the below screen. Select the Alignment tab and select Align with Clustalw.
5. You have the option to modify the parameters of Clustalw, but its fine to keep them on the default. Hit OK and the alignment will run.
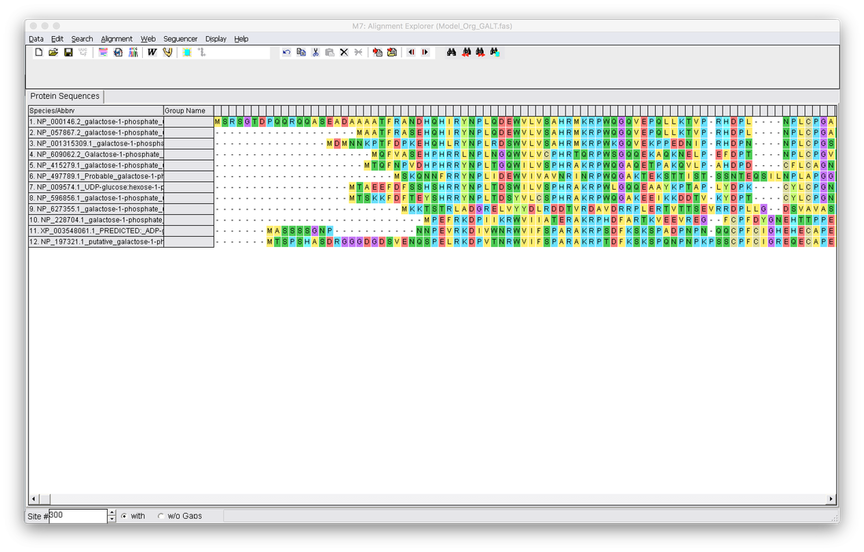
6. Now all your sequences should be aligned. If they are not check to ensure they all are in the correct orientation and that all sequences are homologs. From here you can remove any gaps in the alignment by highlighting the region and then pressing the black X button.
7. Next click on the DATA tab and click on Export Alignment as a FASTA format.
8. Next close the Alignment screen and Click on the Phylogeny tab. Select what type of tree you want to construct (maximum likelihood etc.). Then select your saved alignment file and open it.
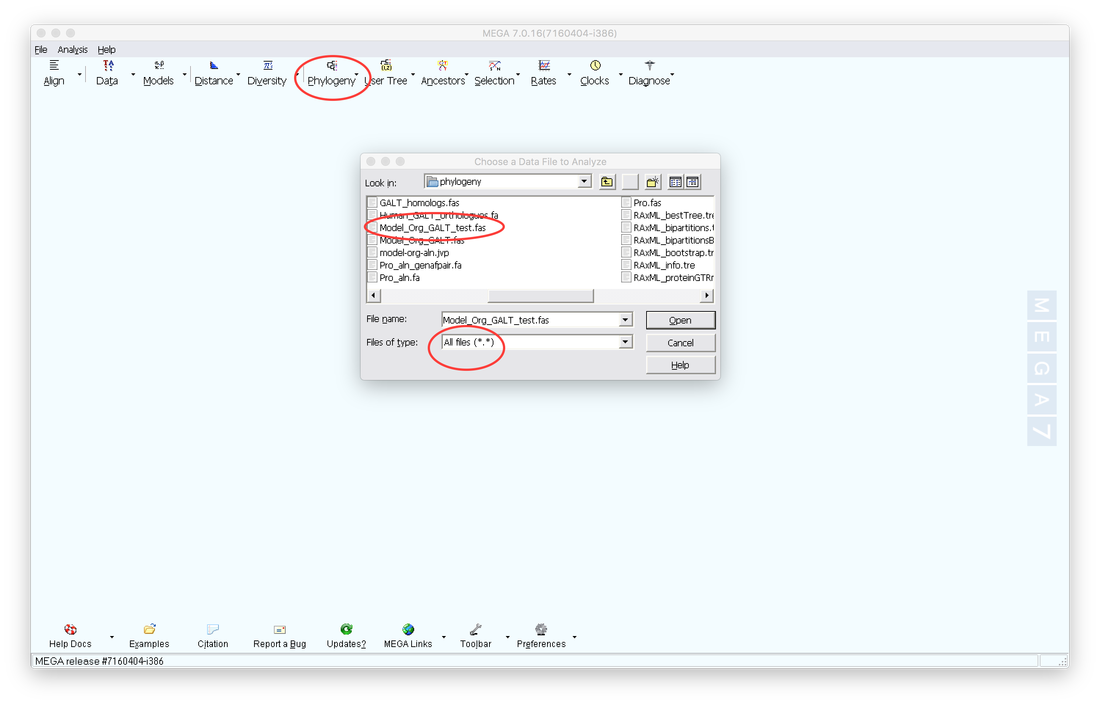
8.
