This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
Introduction
Classic galactosemia is rare disease that is characterized by the inability to process the sugar galactose [1]. Galactosemia is caused by a variety of loss of function mutations in the GALT gene. Phenotypes of galactosemia are manifested in a wide variety of seemingly unrelated symptoms, including: vomiting, diarrhea, cataracts, liver and kidney failure, primary ovarian insufficiency, neurological disorders, and sepsis [2]. Most of these symptoms can be mitigated by following a galactose restricted diet. However, even with a controlled diet many symptoms still occur latter in life with nearly 80% of females developing primary ovarian insufficiency (POI). POI is another name for early menopause, which is characterized by irregular periods, hot flashes, irritability, and infertility. The mechanism behind the cause of these symptoms is largely unknown, but ER stress and abnormal glycosylation have been suggested as possible underlying causes.
|
Still it is unknown how deficiency in the GALT enzyme relates to abnormal glycosylation and POI. To uncover this link this project proposes multiple experiments at better understanding GALT and its role outside of galactose metabolism. GALT encodes a galactose-1-phosphate uridylyltransferase, which catalyzes the second reaction of Lelior pathway (Figure 1). This reaction involves the conversion of UDP-glucose to UDP-galactose, which are critical glycosyl donors for glycosylation reactions [2]. Thus altered levels of both UDP-glucose and UDP-galactose may perturb proper cellular glycosylation. Low levels of glycoproteins are responsible for mis-folded proteins, ER stress, and many alterations of protein receptors. Thus deficiency of the GALT enzyme may cause POI via aberrant glycosylation, therefore even on a galactose restrict diet a patient will inevitably develop this symptom.
|
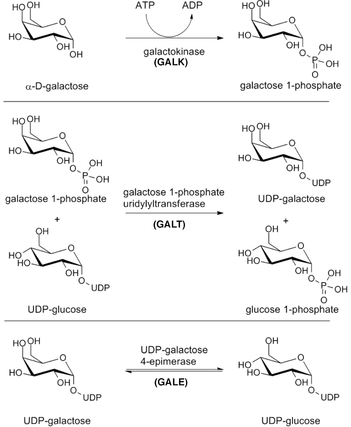
Figure 1. The Lelior pathway of galactose catabolism. |
GALT is extensively connected to many cellular processes, including nucleoside metabolism, glycogen biosynthesis, and protein glycosylation (Figure 2). Thus the expectation that GALT deficiency may lead to abnormal glycosylation is very reasonable. Further, GALT is extremely well conserved protein spanning across all domains of life (Figure 3). Thus, I expect its function and interactions to be conserved in various model organism. I will use the common mouse (Mus musculus) due to its similar disease phenotype to humans and Saccharomyces cerevisiae due to its ease of genetic modification and the ability to screen large numbers of strains quickly. My primary goal is to elucidate how GALT deficiency can lead to premature ovarian insufficiency and to illuminate the broader role of GALT outside its well-known role in galactose catabolism. My hypothesis is that previously overlooked metabolic roles of GALT, including glycosylation, play a central role in the pathology of classic galactosemia.
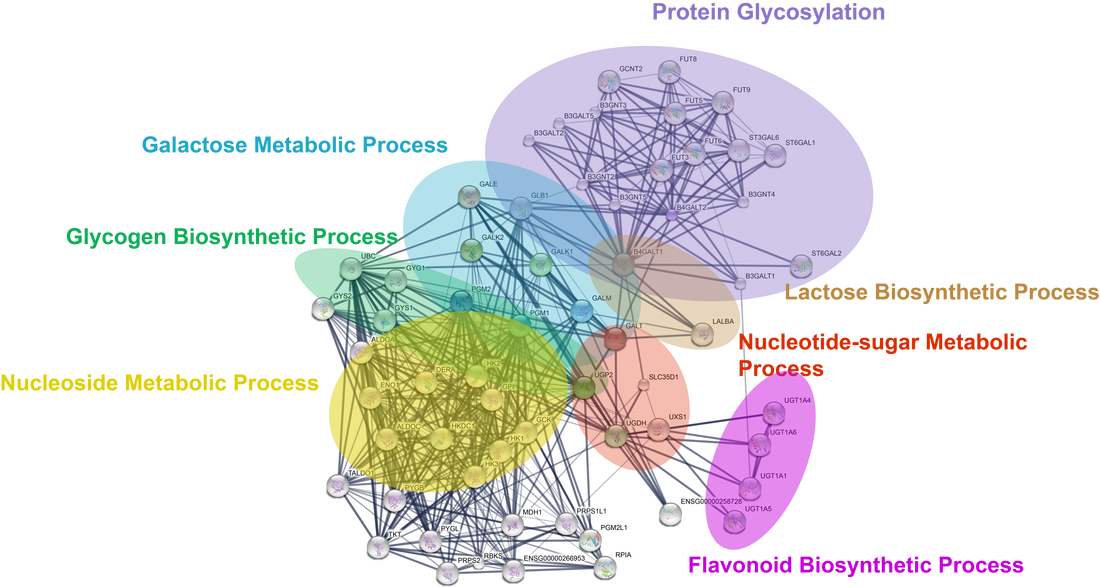
Figure 2. Human GALT protein-protein interaction network. |
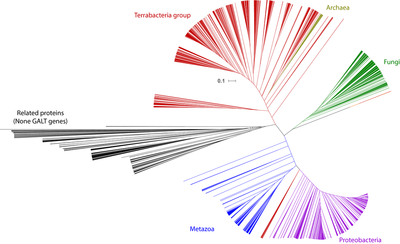
Figure 3. Phylogeny of GALT homologs. |
Specific aim 1
The first aim has two main goals, (1) to link GALT genotype to functionality (ability to process galactose) and (2) linked genotype to the development of POI. I will first screen the functionality of all known Human GALT mutant alleles using a growth assay with S. cerevisiae. Functionality will be defined as the growth on galactose containing media compared to the WT GALT allele. Next, I will select of subset of the Human GALT mutants and use CRISPR/Cas9 to create transgenic mice lines with the various Human GALT disease alleles. I will then screen female mice for those that exhibit premature ovarian insufficiency. Thus in doing so I will be able to predict whether or not a female, after genotyping, can expect to develop POI.

General overview of aim 1. First screen the functionality Human GALT mutant alleles using S. cerevisiae. Next, select a subset of alleles and screen for POI in adult female mice.
Specific aim 2
|
Aim 2 looks to characterize deferentially expressed genes across ovarian development in GALT deficient mice. I will perform RNA-seq on the ovaries of wild type and GALT deficient mice throughout ovarian development and into adulthood, mice will be fed a galactose free diet. RNA-seq data will be sorted using GO terminology and compared between both WT and GALT deficient mice and between the sampled time points. Identifying genes that are deferentially regulated and the time-point this occurs in the ovaries in the absence of GALT are possible targets for identifying novel processes that GALT may modulate and help elucidate the pathology of premature ovarian insufficiency. Since males with galactosemia do not exhibit infertility, I expect gene dysregulation to occur after sex determination happens in development. Further, given the protein interactions of GALT, I expect genes involved in N- and O-glycosylation, ER stress, and various carbon metabolic pathways to be deferentially regulated.
|

Hypothetical gene expression data from RNA-seq experiment. |
Specific aim 3
|
Lastly, aim 3 will address the crux of this project, which is to characterize the ovarian glycoproteome of both WT and GALT deficient mice. I will use the Integrated GlycoProteome Analyzer [5] (IGPA) on ovarian tissue of adult WT and GALT deficient mice. Proteins that have altered glycosylation will be identified by comparing the glycoproteomes of WT vs. GALT deficient mice. Identified proteins will be sorted by their biological process and molecular functions. I expect to see abnormal (hypo, hyper, or newly) glycosylation in the GALT deficient mice. This will lead to ER stress and changes of the glycosylation receptors as the cell deals with defective glycosylation. Identifying proteins that have altered glycosylation in ovarian cells of GALT deficient mice will provide insight to the pathology of premature ovarian insufficiency and other late life effects of galactosemia.
|
Hypothetical results from IGPA. |
Conclusions and future directions
Investigating how GALT deficiency perturbs cellular metabolism via altered glycosylation will hopefully shed light on the obscure pathology of galactosemia and POI. Further, linking GALT genotype to phenotype will allow proper prediction of whether a female patient will develop POI. Since known biochemical markers of galactosemia, namely elevated levels of erythrocyte galactose-1-phosphate and urinary galactitol, do not correlate with the long term outcome of galactosemia discovering a new biomarker is critical. With the data generate via the glycoproteomics we may identify a list of candidate biomarkers. Thus further studies should investigate each to see if they are valid biomarkers for the development of long term outcomes of galactosemia.
