This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
What is phylogenetics
Phylogenetics is the study of evolutionary history and the relationship among biological entities, whether they be individuals, populations species (taxa), or genes. These evolutionary relationships are ascertained through phylogenetic inference methods that use to models of evolution to evaluate either molecular data (DNA, protein) or morphological traits [1]. The output of these inferences are called phylogenies or phylogenetic trees. Phylogenetic trees are composed of few key components: tips (leaves), branches, nodes, clades, a outgroup, and a root (Figure 1).
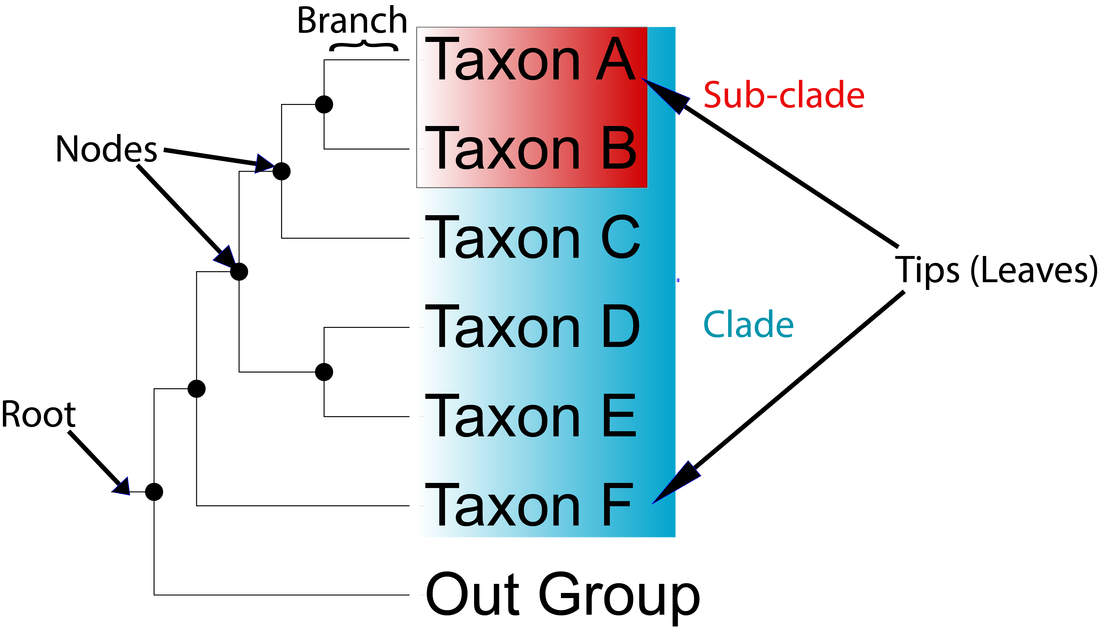
Figure 1. Rooted phylogeny with labeled components
|
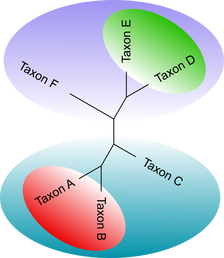
Figure 2. Unrooted phylogeny, monophyletic clades are indicated by colored ovals.
|
The tips or leaves represent the taxa (species) being studied. The lines connecting all the tips are called branches and the point at which they connect are called nodes. Tips that share a common ancestor (node) can be placed into a special group called a monophyletic clade (indicated in Fig 1&2 by colored groups). Some trees have a special leaf called the out group the use of which creates a rooted tree (Fig. 1). Proper outgroups are selected based on prior knowledge of the relationship of the taxa being studied. For example if we are studying a clade of birds we may set an alligator as the out group. If there is insufficient prior knowledge of the relationships and are unable to use an out group, we will generate a unrooted tree (Fig. 2).
For a tutorial on how to use MEGA, RAxML, and MAFFT click HERE!
For a tutorial on how to use MEGA, RAxML, and MAFFT click HERE!
Methods of inferring phylogenies

Figure 3. General work flow of a phylogenetic analysis.
Compiling the Sequences.
The first step in any phylogenetic analysis is to decide what sequences you will need. I went to GenBank and using the HomoloGene tool found protein sequences of homologs of GALT in common organisms. From here I downloaded the sequences into a fasta file, which is plain text file for representing either nucleotide or protein sequences. The fasta file containing the GALT homologs is displayed below.
Sequence Alignment
Once the sequences you wish to analyze have been all put in the same fasta file you must align them. Alignment creates a data matrix out of your provided sequences, where regions of homology between the sequences are aligned [2]. This is a required step to perform a meaningful phylogenetic inference.
There are numerous programs that can be used to align sequence data. Here I use the alignment program MAFFT, which has the advantages of being quick and can handle both small and large data sets [3]. MAFFT utilizes the BLOSUM62 matrix for its amino acid scoring matrix. Briefly, it is a matrix of probabilities describing the likelihood of a biologically meaningful amino-acid residue-pair occurring in an alignment. I then viewed the sequence alignment in the program Jalview, which is displayed below (Fig. 4). As you can see the GALT gene has regions that are conserved across multiple domains of life, indicating these sites are crucial to the enzymatic activity of the protein.
There are numerous programs that can be used to align sequence data. Here I use the alignment program MAFFT, which has the advantages of being quick and can handle both small and large data sets [3]. MAFFT utilizes the BLOSUM62 matrix for its amino acid scoring matrix. Briefly, it is a matrix of probabilities describing the likelihood of a biologically meaningful amino-acid residue-pair occurring in an alignment. I then viewed the sequence alignment in the program Jalview, which is displayed below (Fig. 4). As you can see the GALT gene has regions that are conserved across multiple domains of life, indicating these sites are crucial to the enzymatic activity of the protein.

Figure 4. Global sequence alignment of the GALT gene from various organisms. Residue percent identity is displayed, the darker the blue the more conserved that residue is.
Phylogenetic inference
Phylogenetic inference is the process of estimating the evolutionary relationships among organisms. Below are a few commonly employed methods of phylogenetic inference.
Distance matrix
Distance matrix methods of phylogenetic analysis rely on a simple measure of genetic distance between the sequences. This distance is calculated given a model of either nucleotide or amino acid substitution, with each model having various assumptions of sequence evolution. This constructs a phylogenetic tree which places sequences with the shortest distance between them under the same interior node [1]. In short this method looks to achieve the "shortest" tree. I used the neighbor-joining method to infer the below phylogeny using the program MEGA (Fig. 5). Phylogenetic support was tested with 100 bootstrap replicates.
Advantages: Computationally efficient (very fast, even on extremely large data sets), good for very conserved sequences
Disadvantages: Cannot handle diverged sequences, sensitive to gaps.
Advantages: Computationally efficient (very fast, even on extremely large data sets), good for very conserved sequences
Disadvantages: Cannot handle diverged sequences, sensitive to gaps.
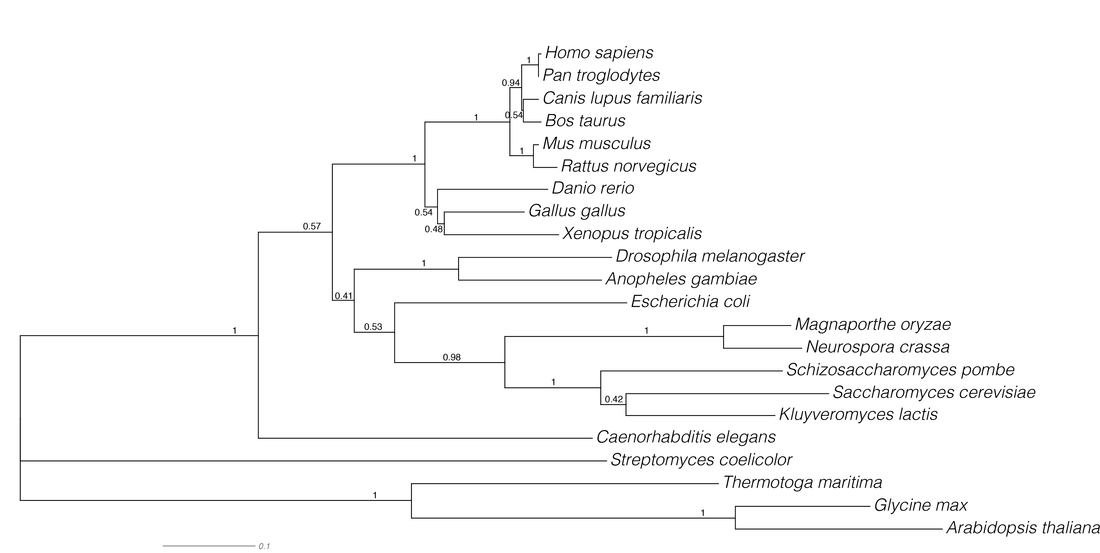
Figure 5. Unrooted neighbor-joining phylogeny. Numbers above branches are bootstrap replicates.
Maximum parsimony
Maximum parsimony is conceptually easy to understand, in short it aims to minimizes the number of changes on a phylogenetic tree by assigning character states to interior nodes [1]. In other terms, the phylogeny which takes the least number of evolutionary events to explain the observed sequence data is the best. Personally, I would never use this method in my own research mainly because it gives no explicit assumptions and thus does not utilize any information of sequence evolution. Again, I use the program MEGA to perform the phylogenetic inference (Fig. 6), running 100 bootstrap replicates.
Advantages: Simplistic (easy to understand)
Disadvantages: Simplistic (no explicit assumptions), susceptible to long branch attraction.
Advantages: Simplistic (easy to understand)
Disadvantages: Simplistic (no explicit assumptions), susceptible to long branch attraction.
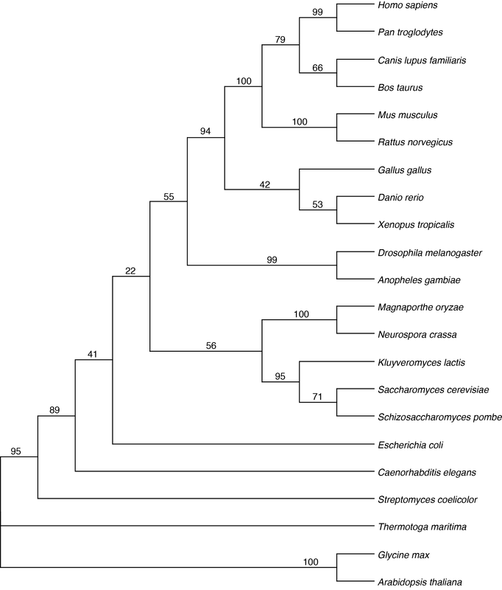
Figure 6. unrooted maximum parsimony phylogeny.
Maximum likelihood
Maximum likelihood phylogenetics is built around the statistical method of likelihood developed by R.A. Fischer [1], defined as the probability of the data given certain parameters. This methods requires a explicit model of sequence evolution and thus trees with more mutations at internodes will have a lower likelihood. This is somewhat similar to parsimony, however likelihood methods allow for independent evolution at sites in the sequence. I used the program RAxML [4] to infer the GALT phylogeny, which is arguable the best implementation of the maximum likelihood method. The below phylogenetic inference (Fig. 7) was performed under a general time reversible protein substitution model with GAMMA model of rate heterogeneity, statistical support was calculated with 1000 bootstrap replicates (bs of 70 roughly corresponds to a p value of 0.05).
Advantages: Explicit and rich repertoire of sophisticated evolutionary models, testable phylogenies
Disadvantages: Computationally demanding, Bootstrap support values are hard to interpret
Advantages: Explicit and rich repertoire of sophisticated evolutionary models, testable phylogenies
Disadvantages: Computationally demanding, Bootstrap support values are hard to interpret
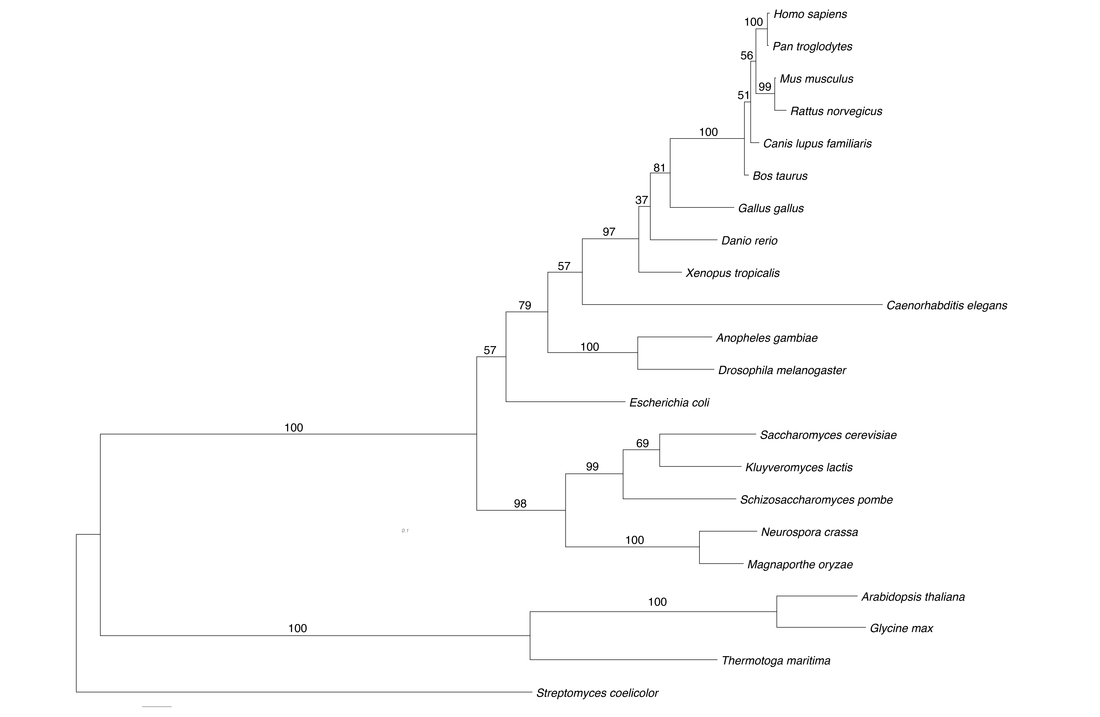
Figure 7. Maximum likelihood phylogenetic inference. Bootstrap support values are displayed above branches.
Bayesian
Bayesian inference is a statistical method where parameters in the model are considered random variables with a certain distributions [1]. Before analysis these parameters are assigned a prior distribution, which is then combined with the data to generate a posterior distribution. However, the posterior probabilities of tree cannot be directly calculated, but algorithms (Markov chain Monte Carlo)
instead sample from this distribution. Each node is given a posterior probability, as with the whole tree, which is simply the probability the node or tree is correct. Bayesian inference was performed using the program MrBayes [5] running 1,000,000 generations using a fixed substitution rate of amino acid evolution (Fig. 8).
Advantages: Sophisticated evolutionary models, testable phylogenies, results are easy to interpret
Disadvantages: Computationally demanding, posterior probabilities tend to be to high, proper prior probabilities are hard to obtain
instead sample from this distribution. Each node is given a posterior probability, as with the whole tree, which is simply the probability the node or tree is correct. Bayesian inference was performed using the program MrBayes [5] running 1,000,000 generations using a fixed substitution rate of amino acid evolution (Fig. 8).
Advantages: Sophisticated evolutionary models, testable phylogenies, results are easy to interpret
Disadvantages: Computationally demanding, posterior probabilities tend to be to high, proper prior probabilities are hard to obtain
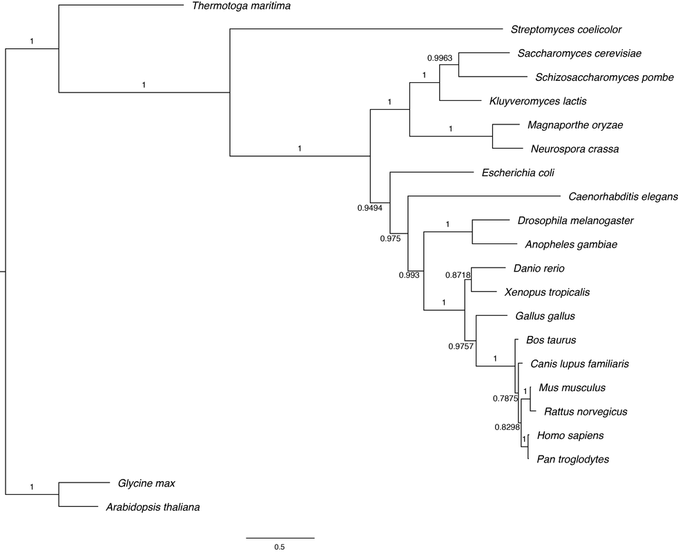
Figure 8. Bayesian phylogenetic inference. Labels above branches are posterior probabilities.
Discussion
The four various methods of phylogenetic inference all arrived at largely similar topologies. As expected the Human GALT gene groups next to the Chimp GALT gene, its most closest relative. However, the most peculiar result of the above phylogenies is that the GALT gene tree is incongruent with the overall species phylogeny (i.e. E. coli clusters within the eukaryote clade). To examine this more closely I ran a preliminary phylogeny using a larger data set. Figure 9 is composed of 4835 proteins including, GALT gene homologs and some closely related proteins, representing taxa spanning across all domains of life. Given the large data set I chose to use a distance matrix (Unweighted Pair Group Method with Arithmetic Mean or UPGMA) to infer the phylogeny and no bootstrapping was performed. Due to the lack of statistical support, simple model, and perhaps not dense enough taxon sampling, this result should be taken with a hefty amount of skepticism until a more robust phylogeny is made.
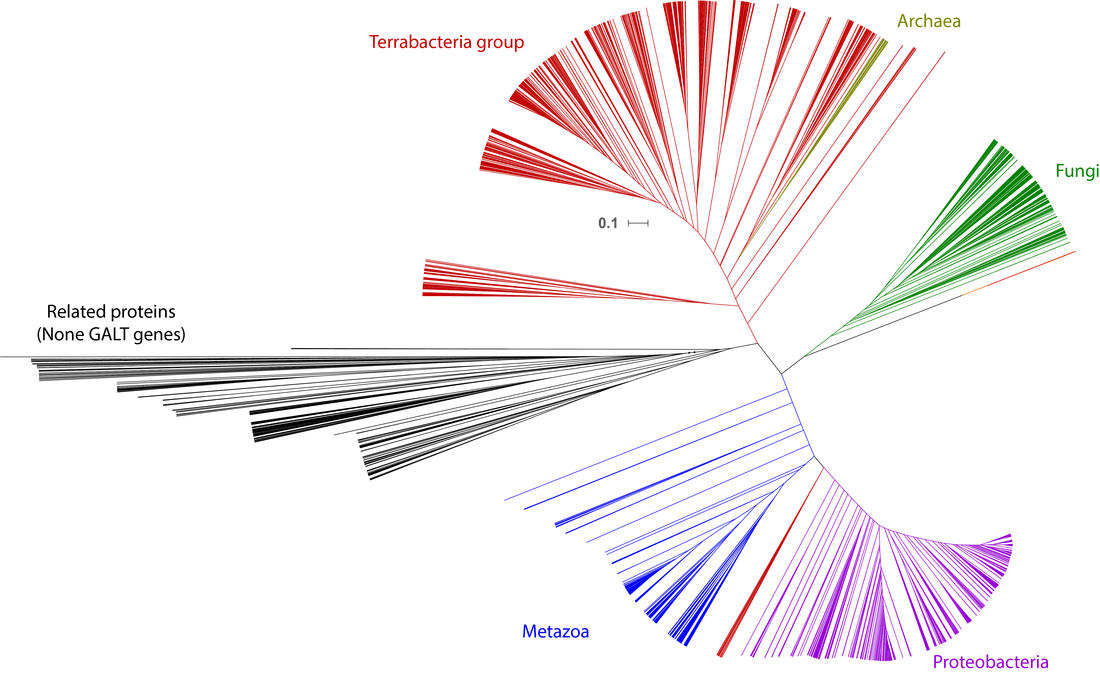
Figure 9. Average linkage phylogeny displays unexpected topologies between archaea, prokaryotes, and eukaryotes.
