This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
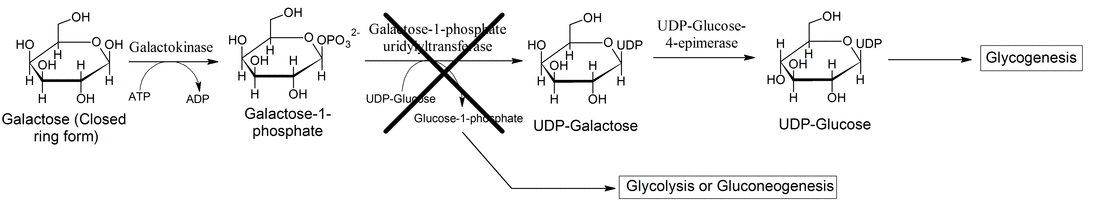
Fig. 1. The Leloir pathway of galactose metabolism. The step GALT catalyzes is crossed out.
What is galactosemia?
|
Galactose metabolism, commonly referred to as the Leloir Pathway, was elucidated in the 1940's by Luis Federico Leloir and colleagues, which won him the Noble Prize for Chemistry in 1970. This pathway and the enzymes involved are highly conserved across Eukaryotes and Prokaryotes. In the yeast model system deficiency of these enzymes results in a negative growth phenotype, when grown on galactose. In Humans, deficiency in these enzymes results in the rare metabolic disease galactosemia, which causes an inability to break down the simple sugar galactose [1]. Hence, as the name implies, galactose accumulates in the blood stream. There are three types of galactosemia, Type I, Type II, and Type III, each caused by a deficiency in a different enzyme involved in the breakdown of galactose. Type I, or classic galactosemia, is the most severe and common - affecting 1 in 30,000 to 60,000 births. However, the disease rate varies among Human populations, being strikingly common among Irish Traveler populations, reaching frequencies near 1 in 450 births. [2].
In 1917 a German physician, Fredick Goppert, first reported the disease [3]. Subsequently, in 1953 the genetic cause of the disease was first demonstrated to be linked a defect in galactose metabolism [4]. Classic galactosemia is caused by a deficiency (mutation) in the enzyme galactose-1-phosphate uridylyltransferase (GALT; EC 2.7.7.12) , which is the second enzyme in galactose metabolism [5]. In individuals with a mutated GALT gene the Lelior pathway is blocked, thus galactose builds up and reaches toxic levels in the blood stream. |
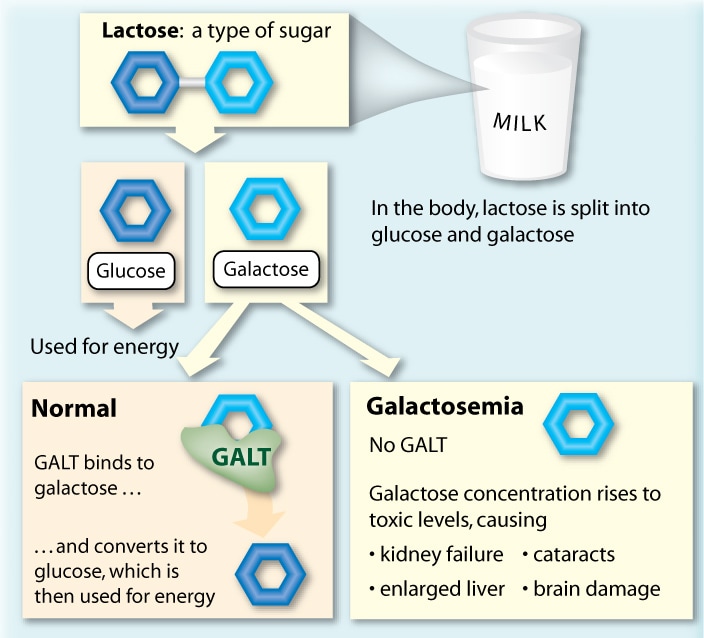
Fig 2. Overview of the main source of galactose and the underlying cause of the disease.
|
What causes galactosemia?
|
Classic galactosemia is inherited in an autosomal recessive fashion, meaning both copies of the gene need to be defective for symptoms to surface [6]. Carriers, or those with a functional and a dysfunctional copy of the gene GALT, produce enough of the GALT enzyme to avoid any symptoms of classic galactosemia. Almost all reported cases of classic galactosemia result from near complete elimination in function of the GALT gene product. More than 250 disease variants of the GALT gene have been recorded in 24 different populations worldwide [7]. The three most common disease variants found are Q188R, S135L, and K285N making up 49%, 7%, and 4%, respectively [8]. These three disease variants appear to be disproportionally distributed among Human populations and ethnicities. For example variant Q188R is reported to account for 70% of disease variants in Caucasians and 0% of disease variants in Japanese [9]. Whereas, variant S135L is found primarily in peoples of African decent [10].
|
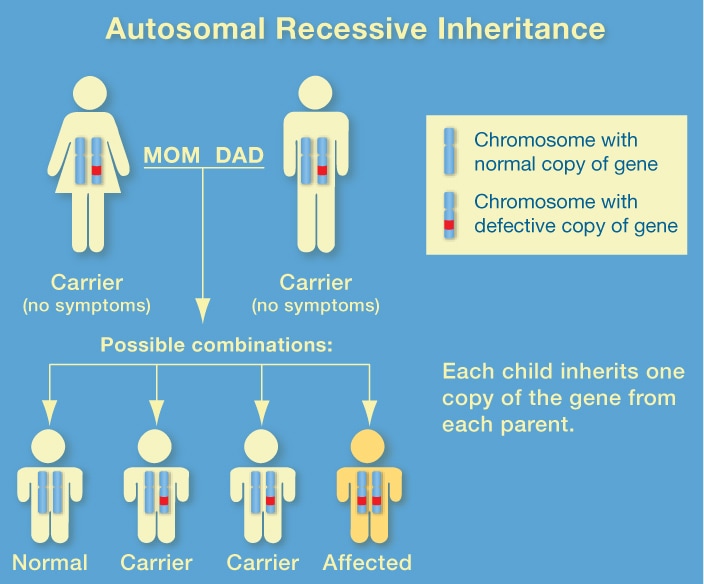
Fig. 3. Inheritance pattern of a GALT disease variant.
|
Gene and protein function
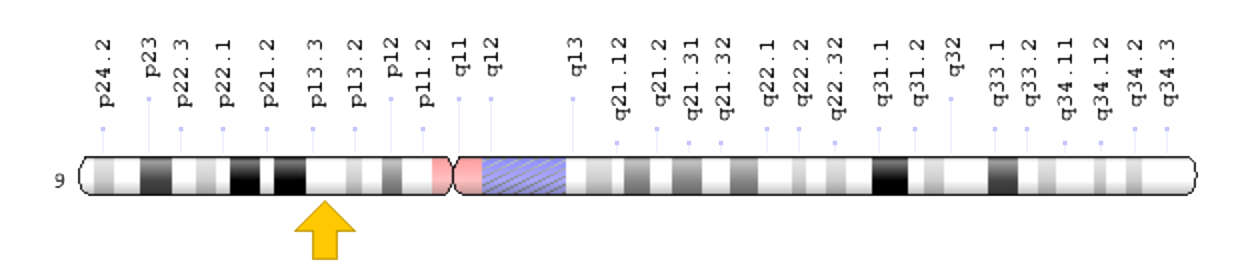
Fig. 4. Human Chromosome IX. Yellow arrow indicates GALT at locus 9p13.3
|
The human GALT gene is located on the short arm (p) of chromosome 9. When GALT functions properly it converts UDP-glucose and galactose 1-phosphate to glucose 1-phosphate and UDP-galactose [16]. Glucose 1-phosphate then enters glycolysis and is metabolized. UDP-galactose is converted back to UDP-glucose by another enzyme (GALE) and is used again in the above reaction [16].
|
Symptoms of classic galactosemia
|
Galactosemia can manifest itself in a variety of afflictions, including kidney failure, liver damage, cataracts, abnormal bleeding, jaundice, failure to thrive, and intellectual disabilities [1],[11]. Serious complications may also arise, which include overwhelming bacterial infections (sepsis), shock, and death [1]. Newborns are most vulnerable and are immediately susceptible to these life-threatening complications if not put on a galactose free diet (removal of ingesting breast milk or lactose-containing formulas) [11]. Infants who survive the neonatal period and continue to ingest galactose may develop serious brain damage [12]. In addition, females may develop reproductive problems, caused by premature ovarian insufficiency [1].
|
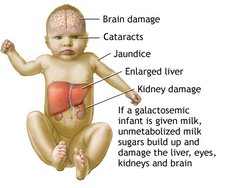
Fig. 5. Common symptoms of galactosemia if a galactose restricted diet is not followed.
|
Prevention and treatment of classic galactosemia
The most pertinent measure in reducing the likelihood of symptoms from manifesting is to implement newborn screening within 10 days of birth, failure to do so is catastrophic on the prognosis. If a galactose free diet is implemented within days of birth, the symptoms quickly abate and prognosis is good [11]. However, even with early and sufficient therapy, long-term outcome into adulthood is variable and typical symptoms may arise [13]. The quality of life in adults with classic galactosemia is overall reduced. Data on long-term outcomes indicates that complications do not correlate with any well known biochemical predictors (galactose-1-phosphate levels). Furthermore, manifestation of these complications vary among individuals, even those with the same genotype [14].
While the long-term prognosis may be variable, prenatal and especially newborn screening are key in identifying those affected and promptly administrating a proper treatment. To measure the extent of the disease in newborns with classic galactosemia, the following is recommended [15].
While the long-term prognosis may be variable, prenatal and especially newborn screening are key in identifying those affected and promptly administrating a proper treatment. To measure the extent of the disease in newborns with classic galactosemia, the following is recommended [15].
- Consultation with a specialist in biochemical genetic disorders
- Measurement of erythrocyte galactose-1-phosphate concentration and urinary galactitol as a baseline for monitoring the effect of treatment
- Neurologic examination and brain MRI as needed
- Ophthalmologic examination, including slit lamp examination for cataracts
- Evaluation for hepatocellular disease, especially in patients with late-treated disease who may be at risk for cirrhosis
Resources

The Galactosemia Foundation
|
Children Living with Inherited Metabolic Diseases (CLIMB)
|
Save Babies Through Screening Foundation, Inc.
|
